2019年7月11日,amjs澳金沙门线路李力课题组,医学院附属仁济医院薛婧课题组和孙勇伟课题组,在GUT杂志上发表了题为Loss of Setd2 promotes Kras-induced acinar-to-ductal metaplasia and epithelia-mesenchymal transition during pancreatic carcinogenesis的研究工作,证实了SETD2缺失协同KRAS促进胰腺癌发生发展,并阐明了SETD2在胰腺癌起始和转移两个阶段的不同功能和机制。
胰腺癌被称为“癌中之王”,恶性程度高、发展和恶化速度快,确诊时多为晚期,且至今没有特别有效的治疗方法,从而具有较高的死亡率。因此针对其发病机制的研究极为迫切。90%以上的胰腺癌由外分泌细胞产生,即为胰腺导管腺癌(Pancreatic ductal adenocarcinoma, PDAC)。起始和转移是PDAC发生发展过程中最重要的两个阶段。炎症和Kras突变均可导致腺泡导管化生(Acinar to Ductal Metaplasia,ADM),且在Kras持续激活的情况下不可逆进展。目前ADM被认为是PDAC起始的重要事件。在抑癌基因失调或炎症、毒性等刺激的情况下,癌前病变的导管样细胞会进一步进展为PDAC。胰腺肿瘤容易发生远端转移,是胰腺癌患者死亡的主要原因。上皮间质化(Epithelial-Mesenchymal Transdifferentiation,EMT)是癌细胞转移的第一事件。表观调控异常是恶性肿瘤的重要特征之一,近年来已发现胰腺肿瘤中也大量存在表观修饰异常,但相关机制和功能研究仍比较欠缺。
组蛋白甲基转移酶SETD2(SET domain-containing protein 2),是目前已知H3K36me3形成的唯一三甲基化酶。目前已知SETD2/H3K36me3主要功能包括参与DNA损伤修复,维持染色质活跃状态、协助转录延伸从而促进基因转录水平。血液肿瘤、胃肠道间质瘤、结肠癌、神经胶质瘤、乳腺癌等中均发现SETD2的突变和其抑癌功能,可见SETD2-H3K36me3是肿瘤中常见的一种抑癌机制,但调控机制具有肿瘤组织的特异性。已有二代测序发现胰腺癌患者同样携带一定比例的SETD2突变,然而其在胰腺肿瘤中的功能和机制尚未明确。
李力副研究员在2014年建立了Setd2条件性基因敲除小鼠模型并在生殖、发育和癌症等领域展开广泛的功能研究(Zuo et al, J Bio Chem, 2018;Wang et al, PLoS Biol, 2018;Xu et al, Nat Genet, 2019)。合作组由此展开进一步研究,结合共用数据库和临床样本,发现SETD2和H3K36me3表达与胰腺癌患者预后显著负相关。通过构建胰腺特异性敲除Setd2,以及联合Kras突变的小鼠,发现SETD2缺失可以大大促进KRAS诱导的胰腺癌进程。分别对腺泡细胞以及胰腺癌细胞Setd2野生型和Setd2缺失的样本进行高通量的表达谱和H3K36me3 ChIP联合分析,发现两个病理阶段中Setd2通过调控不同下游基因而发挥作用:1)起始阶段-调控MYC蛋白的E3泛素连接酶Fbxw7表达,影响腺泡细胞稳态,从而促进ADM发生;2)转移阶段:调控Ctnna1粘连蛋白的表达,影响E-cadherin稳定性,从而促进EMT及转移 。
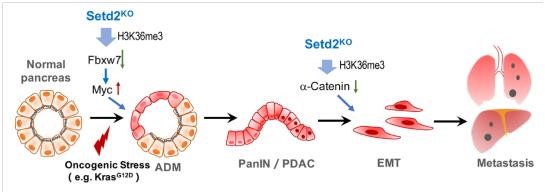
不同病理时期的差异性调节机制,赋予Setd2在复杂疾病中的不同生物学功能,从而更加精准的调控病理过程。本研究首次揭示了SETD2-H3K36me3表观调控元件协同Kras突变在胰腺癌中的功能与分子机制,深入了我们对其病理机制的认识,为胰腺癌靶向治疗提供了新的思路。
牛宁宁副研究员和路平博士为该论文的共同第一作者;薛婧研究员、李力副研究员和孙勇伟主任为该论文的共同通讯作者。
原文链接:









